Rated 4.7 out of 5 stars
4.7 / 5
















726,000+ customers
Free 60-Day Returns
1-Year Warranty
726,000+ customers
Free 60-Day Returns
1-Year Warranty

You're shopping for a skincare device and see "FDA-Cleared" on the packaging. That sounds official, legitimate, safe. But then you notice another product claiming "FDA-Approved" status. Wait, are those the same thing? Is one better than the other? Did the first product fail to get full approval?
The confusion is understandable and widespread. These terms sound similar, appear interchangeably in marketing materials, and most consumers reasonably assume they mean roughly the same thing. They don't. Understanding the difference between FDA-cleared and FDA-approved matters for making informed decisions about the devices you use on your skin.
Before diving into the specific distinctions, it helps to understand why the FDA uses different regulatory pathways at all.
The Food and Drug Administration oversees an enormous range of products: prescription medications, over-the-counter drugs, medical devices, food, cosmetics, and more. Each category presents different risks and requires different evaluation approaches.
A new cancer drug that alters fundamental biological processes requires different scrutiny than a bandage that covers wounds. A pacemaker implanted in someone's chest needs more rigorous evaluation than a digital thermometer. The FDA developed different regulatory frameworks to match the level of oversight to the actual risk involved.
For medical devices specifically, the FDA categorizes products into three classes based on risk level, and each class follows different regulatory requirements.
Class I devices present minimal risk and include products like bandages, tongue depressors, and non-powered examination gloves. Most Class I devices are exempt from premarket review entirely, though they must still meet general manufacturing standards.
Class II devices present a moderate risk and include products like powered wheelchairs, pregnancy tests, and many skincare devices. These require premarket notification, which is where "clearance" enters the picture.
Class III devices present the highest risk, typically because they support or sustain life, are implanted, or present a potential unreasonable risk. Examples include pacemakers, breast implants, and replacement heart valves. These require the most rigorous pathway: premarket approval.
FDA approval specifically refers to the Premarket Approval (PMA) process, the most stringent regulatory pathway for medical devices.
When a manufacturer wants to bring a Class III device to market, they must submit a Premarket Approval application. This requires extensive clinical data demonstrating that the device is safe and effective for its intended use.
The process involves laboratory testing and often animal studies, human clinical trials conducted under strict protocols, detailed manufacturing information, proposed labeling review, and FDA inspection of manufacturing facilities.
According to FDA documentation, the PMA process requires "valid scientific evidence" that provides "reasonable assurance" of safety and effectiveness. This evidence typically comes from well-controlled clinical investigations.
The review process takes approximately 180 days by statute, though actual timelines often extend longer. The FDA conducts a thorough evaluation of all submitted data before making a decision.
When a device receives FDA approval, it means the FDA has reviewed extensive clinical evidence supporting the device's safety and effectiveness. The agency has determined, based on this evidence, that the device can be legally marketed for its approved indications.
Approval represents the highest level of FDA endorsement for a medical device. The manufacturer proved, through rigorous clinical trials, that the device works as claimed and doesn't present unreasonable risks.
FDA approval is reserved for high-risk devices where failure could have serious consequences. Examples include implantable cardiac devices like pacemakers and defibrillators, artificial joints for hip and knee replacement, surgical mesh for hernia repair, and certain diagnostic devices where misdiagnosis could be dangerous.
Most skincare devices don't fall into this category. They're not implanted, they don't sustain life, and their risk profile doesn't warrant the Class III designation that triggers the PMA requirement.
FDA clearance refers to the 510(k) premarket notification process, the regulatory pathway for most Class II medical devices, including many skincare devices.
The 510(k) pathway has a fundamentally different logic than PMA. Rather than proving a device is safe and effective through extensive clinical trials, the manufacturer demonstrates that their device is "substantially equivalent" to a legally marketed device that already exists.
This predicate device, as it's called, serves as the comparison point. The manufacturer shows that their new device has the same intended use, has the same technological characteristics or different characteristics that don't raise new safety questions, and is at least as safe and effective as the predicate.
The process involves technical documentation of device specifications, comparison to the predicate device, performance testing data, biocompatibility information where relevant, and proposed labeling review.
Clinical data may or may not be required depending on the specific device and how different it is from the predicate. Many 510(k) submissions rely primarily on bench testing and engineering data rather than human clinical trials.
When a device receives 510(k) clearance, it means the FDA has determined the device is substantially equivalent to an existing legally marketed device. The agency has reviewed the comparison and agrees that the new device doesn't present significantly different safety or effectiveness concerns.
Clearance indicates the device has passed FDA review and can be legally marketed. It represents meaningful regulatory oversight, just through a different framework than approval.
The 510(k) pathway applies to moderate-risk devices that have predicates already on the market. This includes most powered skincare devices like LED therapy devices and galvanic devices, many diagnostic devices, surgical instruments, and numerous other medical devices that present moderate but not high risk.
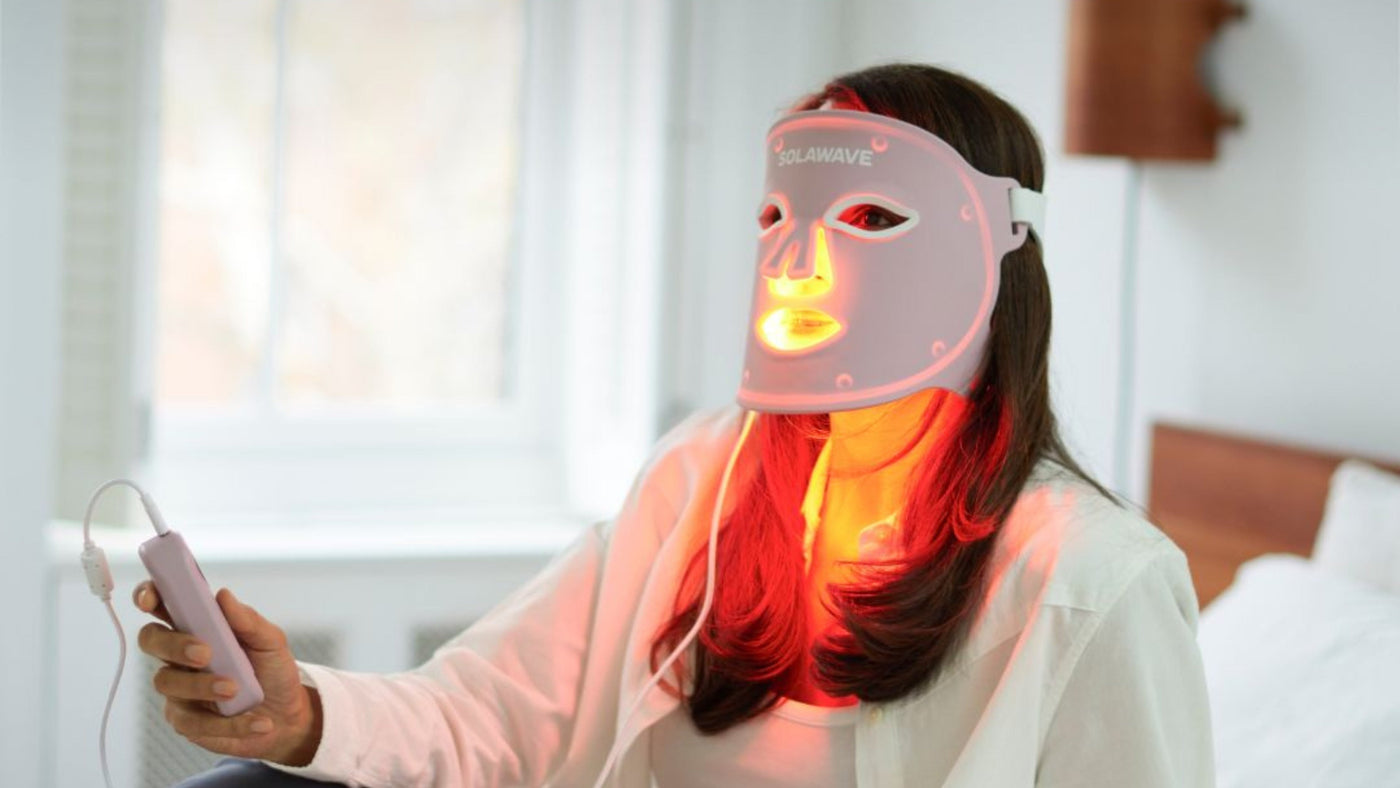
The Red Light Therapy Face Mask is FDA-cleared, meaning it has completed the 510(k) process and demonstrated substantial equivalence to a predicate device. This represents appropriate regulatory oversight for an LED-based skincare device.
Now you can see why these terms aren't interchangeable.
FDA approval requires extensive clinical trials demonstrating safety and effectiveness. Manufacturers invest years and millions of dollars generating this evidence before submission.
FDA clearance requires demonstration of substantial equivalence to a predicate device. Clinical data may be required in some cases, but the core requirement is showing comparability rather than proving efficacy from scratch.
FDA approval addresses high-risk devices where failure could cause serious harm. The extensive evidence requirement matches the serious potential consequences.
FDA clearance addresses moderate-risk devices where existing safety profiles of similar devices provide reasonable assurance. The substantial equivalence standard makes sense because similar devices have already been used safely.
FDA approval typically takes 180 days or longer due to the extensive evidence review required.
FDA clearance typically takes 90 days, though timelines vary based on submission complexity and FDA workload.
FDA approval is significantly more burdensome for manufacturers, requiring extensive clinical programs and larger submissions.
FDA clearance, while still substantial, involves less extensive documentation and a faster pathway to market.
Both pathways result in legally marketable devices that have passed FDA review. The difference lies in that review, not its legitimacy.
Several misunderstandings persist about these regulatory pathways.
This assumes that a less rigorous review means lower safety standards. In reality, the 510(k) pathway is appropriate for devices where existing safety profiles of similar devices provide confidence. A new LED therapy device doesn't need novel clinical trials when LED therapy devices have been used safely for years.
The clearance pathway acknowledges that not every device needs de novo evidence when substantial experience with similar devices already exists.
For high-risk devices, approval provides appropriate reassurance. For moderate-risk devices like most skincare technology, clearance is the appropriate pathway. An LED face mask seeking approval would be pursuing the wrong regulatory strategy, not a higher safety standard.
Neither clearance nor approval means the FDA has verified specific marketing claims beyond the device's intended use. Manufacturers must still comply with regulations about claims, but the FDA doesn't review every marketing statement.
Some skincare devices fall outside FDA jurisdiction entirely if they don't make medical claims. A device marketed purely for cosmetic purposes without therapeutic claims might not go through either pathway. Clearance indicates the device does make medical claims and has undergone appropriate review.
Understanding the difference between FDA-cleared and FDA-approved helps you evaluate the devices you're considering.
When a skincare device is FDA-cleared, you know the manufacturer submitted the device for FDA review rather than avoiding regulatory oversight. You know the FDA determined the device is substantially equivalent to a predicate that's already legally marketed. You know the device meets applicable performance and safety standards. And you know the device can legally make the therapeutic claims in its labeling.
This represents meaningful regulatory oversight appropriate for the device category.
FDA clearance is the standard you should expect for LED therapy devices, galvanic devices, and other powered skincare technology making therapeutic claims. Devices without any FDA status may be avoiding regulatory oversight by making only cosmetic claims, may be new to market and still pursuing clearance, may be international products not yet submitted to the FDA, or may be making claims outside their regulatory scope.
The red light therapy wand combines red light with galvanic, therapeutic warmth, and galvanic current. The wand requires a water-based serum like the LightBoost Activating Serum to activate all four technologies.
When evaluating skincare devices, consider asking whether the device is FDA-cleared, what the clearance covers (which claims are actually reviewed), and whether the manufacturer can provide the 510(k) clearance number.
Legitimate manufacturers are transparent about their regulatory status because it represents a competitive advantage over non-cleared alternatives.
Regulatory clearance is important, but not the only factor in evaluating skincare devices.
FDA clearance requires compliance with the Quality System Regulation (QSR), ensuring consistent manufacturing standards. However, quality varies even among cleared devices. Look for manufacturers with established reputations and transparent quality practices.
While clearance doesn't require clinical trials, some manufacturers conduct them anyway. Published research supporting a device's claimed benefits provides additional confidence beyond regulatory status alone.
A dermatologist's recommendation indicates professionals who understand skin biology believe the device provides value. This represents an independent evaluation beyond what regulatory clearance provides.
All Solawave devices are FSA/HSA eligible and recommended by dermatologists, indicating both regulatory compliance and professional endorsement.
Established companies with track records of quality products present a lower risk than unknown manufacturers. Customer service responsiveness, warranty policies, and how companies handle issues all matter.
Understanding FDA regulation helps you make informed device choices, but devices work best as part of complete routines.
FDA-cleared devices enhance but don't replace topical skincare. The LightBoost Niacinamide Face and Neck Serum provides barrier support and tone improvement that complements device treatment.
The LightBoost Face and Neck Cream delivers intensive hydration to support overall skin health alongside device benefits.
The red light therapy eye mask addresses the delicate eye area, while the LightBoost Collagen Caffeine Eye Cream provides complementary topical support.
The Neck & Chest Rejuvenating Mask extends device treatment to commonly neglected areas that show aging prominently.
Whatever devices and products you choose, consistent use produces results. The most sophisticated FDA-cleared device provides minimal benefit if used sporadically.
The difference between FDA-cleared and FDA-approved isn't about safety levels or regulatory rigor in the way most consumers assume. These terms describe different regulatory pathways appropriate for different device risk profiles. FDA approval, reserved for high-risk devices, requires extensive clinical trials proving safety and effectiveness. FDA clearance, used for moderate-risk devices including most skincare technology, requires demonstrating substantial equivalence to existing legally marketed devices. For skincare devices, clearance is the appropriate standard. It indicates meaningful FDA review and legal marketability without the burden of novel clinical trials that the device category doesn't warrant. When you see "FDA-Cleared" on a skincare device, you're seeing evidence of appropriate regulatory compliance, not a lesser standard than approval. Understanding FDA-cleared vs FDA-approved empowers you to evaluate device claims accurately and make informed purchasing decisions based on what these terms actually mean.
Ready to experience FDA-cleared skincare technology? Shop Solawave's skincare collection today.
No. These terms describe different regulatory pathways. FDA approval requires extensive clinical trials and applies to high-risk devices like pacemakers. FDA clearance requires demonstrating substantial equivalence to existing devices and applies to moderate-risk devices like most skincare technology. Both result in legally marketable devices that have passed FDA review, but through different processes appropriate to different risk levels.
Most skincare devices don't meet the criteria for the Class III designation that triggers the approval requirement. They're not implanted, don't sustain life, and don't present the high-risk profile that warrants extensive clinical trials. Clearance is the appropriate pathway for these moderate-risk devices. Seeking approval would mean pursuing an unnecessarily burdensome pathway.
FDA clearance indicates the device has passed FDA review and met applicable safety standards for its category. It means the device is substantially equivalent to an existing legally marketed device with an established safety profile. No regulatory status guarantees absolute safety, but clearance represents meaningful oversight and legal marketability.
Yes. The FDA maintains a 510(k) database where you can search for cleared devices by manufacturer, device name, or clearance number. Legitimate manufacturers can provide their 510(k) clearance number, and you can verify it through the FDA's public database at accessdata.fda.gov.
Devices without FDA clearance may be making only cosmetic claims that don't require FDA oversight, may be international products not submitted to the FDA, may be new products still pursuing clearance, or may be improperly marketed without required regulatory compliance. For devices making therapeutic claims about changing skin structure or function, FDA clearance is expected. Its absence warrants questions about the product's regulatory status.